
Bayesian phylogenetic analysis
Summary
Bayesian statistics is named after Thomas Bayes, a presbyterian priest and amateur mathematician who lived in the 18th century. He showed how to "invert" probabilities, so that you can calculate the likelihood that a hypothesis is correct. In standard statistical inference, one is forced to address this problem indirectly.
Bayes's method led to difficult mathematical equations that could not be solved with pen and paper. However, in the 1950s, scientists in the US nuclear weapons program developed effective numerical methods to solve these problems. In combination with today’s powerful computers, Bayesian statistics have now become a standard method in most scientific disciplines.
We were among the first to develop software and algorithms for Bayesian inference of evolutionary trees. Today, evolutionary trees – or phylogenies, as they are also called - are usually estimated from genetic data. The applications of phylogenetic analysis are surprisingly varied. They include tracking of viral transmission pathways, estimation of when and where different groups originated, and reconstruction of the relations between different languages, just to name a few examples.
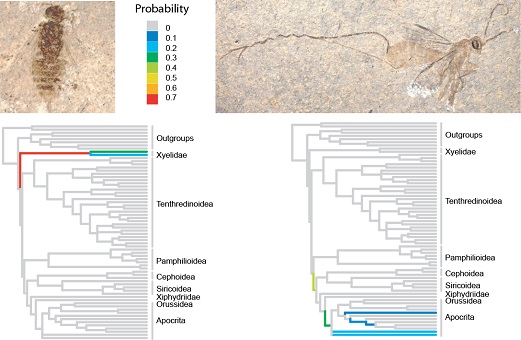
Our work is largely done within the context of the software packages MrBayes and RevBayes. Among other things, we are interested in how evolutionary models can be formulated in a generic way, and we are trying to improve the numerical methods used in Bayesian phylogenetic inference. We also work with several different applications in evolutionary biology, for example, understanding how morphological characters change over time and how fossils can be used in dating past evolutionary events.
Links
MrBayes (http://mrbayes.net )
)
RevBayes (http://revbayes.com)
The research is supported by the Swedish Research Council (VR)
Project Participants at NRM
Fredrik Ronquist
Allison Hsiang
Johan Nylander
Selected Publications
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard M, Huelsenbeck JP. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542.
Ronquist F, Klopfstein S, Vilhelmsen L, Schulmeister S, Murray D, Rasnitsyn AP. 2012. A total-evidence approach to dating with fossils, applied to the early radiation of Hymenoptera. Systematic Biology 61: 973–999.
Höhna S, Heath T, Boussau B, Landis M, Ronquist F, Huelsenbeck JP. 2014. Probabilistic graphical model representation in phylogenetics. Systematic Biology 63: 753-771.
Klopfstein S, Vilhelmsen L, Ronquist F. 2015. A nonstationary Markov model detects directional evolution in hymenopteran morphology. Systematic Biology 64: 1089–1103.
Zhang C, Stadler T, Klopfstein S, Heath TA, Ronquist F. 2016. Total-Evidence Dating under the Fossilized Birth-Death Process. Systematic Biology 65: 228–249.
Ronquist F, Lartillot N, Phillips MJ. 2016. Closing the gap between rocks and clocks using total-evidence dating. Philosophical Transactions of the Royal Society of London B 371: 20150136.
Lartillot N, Ronquist F, Phillips MJ. 2016. A mixed relaxed clock. Philosophical Transactions of the Royal Society of London B 371: 20150132.
Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F. 2016. RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Systematic Biology 65: 726–736.